урок Школы Видеоблогера. ‒ #100по100
Как скопировать ссылку с интернета на свой профиль?
Для того чтобы скопировать ссылку на свой профиль в ВК, заходите в вк и затем смотрите строку браузера, то есть, когда вы просто зашли на главную страницу, например, нажали на логотип ВК, то у вас здесь написано feed, и не важно, что вы в своем аккаунте, все равно эта ссылка не на ваш профиль.
Чтобы получить ссылку именно на ваш профиль, необходимо:
- нажать «моя страница»,
- копировать ссылку правой клавишей мышки,
- и вставить ссылку в анкету или куда вам необходимо ее прописать.
Чтобы прислать ссылку на свой профиль в facebook, необходимо:
- зайти на Facebook (на главной странице эта ссылка не на ваш профиль),
- вверху нажать на свой логотип с именем,
- копировать ссылку, перейдя в свой профиль, правой клавишей мышки,
- и вставить туда, куда вам нужно (например, в анкету).
Чтобы скопировать ссылку на свой профиль в Одноклассниках, сначала вы:
- заходите под своим логином и паролем в Одноклассники,
- (даже если вы видите страницу, как будто бы это ваша, обратите внимание на адресную строку, где одноклассники.
 ру написано feed, то есть это ссылка не на ваш профиль),
ру написано feed, то есть это ссылка не на ваш профиль), - нажмите на свою фотографию и затем выбрать имя,
- ссылка на ваш профиль появится в адресной строке,
- копируйте ссылку правой кнопкой мыши и вставляйте в анкету.
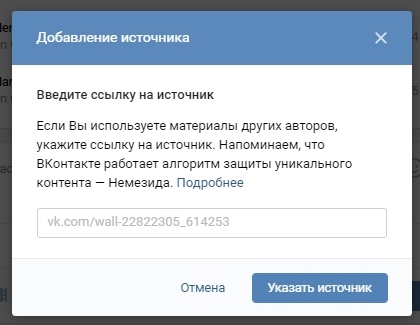 ру написано feed, то есть это ссылка не на ваш профиль),
ру написано feed, то есть это ссылка не на ваш профиль), Чтобы предоставить ссылку на свой профиль в Instagram:
- заходите в Instagram в свой профиль через компьютер,
- находите иконку человека, и нажимаете,
- и из адресной строки правой клавишей мыши, копировать ссылку.
Чтобы скопировать свой логин в skype, по которому вас можно будет добавить:
- заходите в skype,
- нажмите на свою фотографию,
- в выпадающем меню выберите профиль skype,
- и здесь есть логин skype. Именно его вы копируете и вставляете в анкету.
Как сделать ссылку на Инстаграм в ВК
Разработчики современных социальных сетей стараются связывать и интегрировать их друг с другом для удобства использования.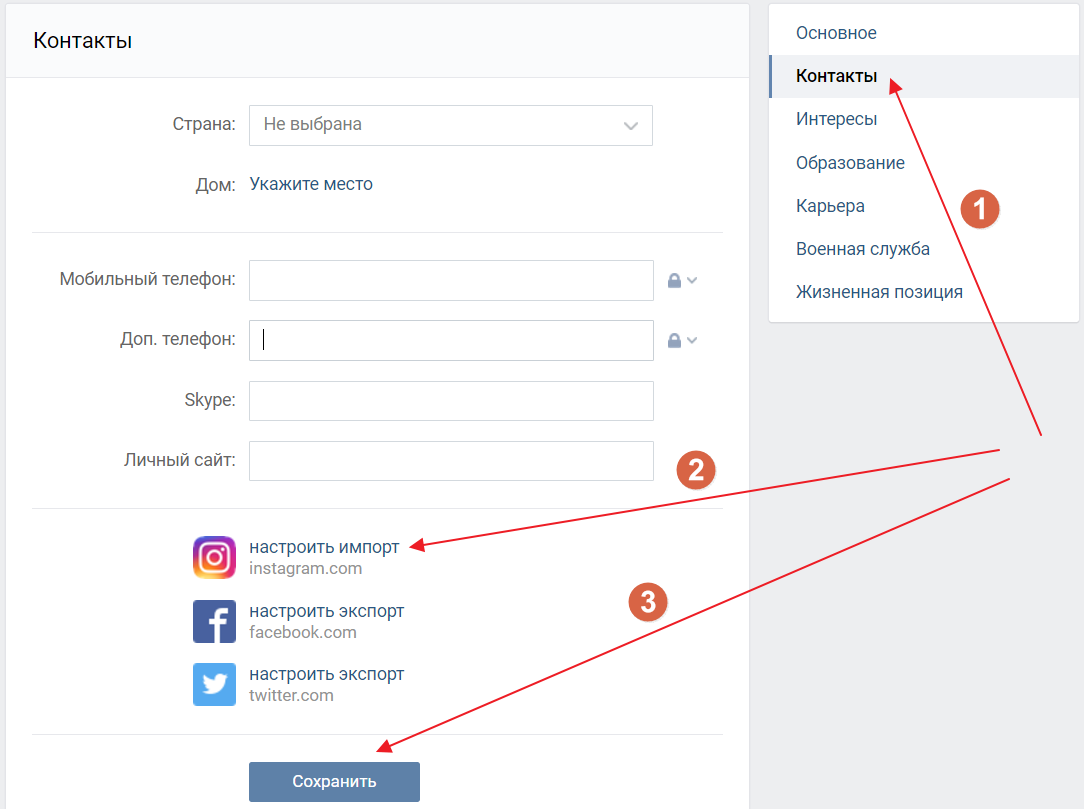 Например, пользователи Instagram могут синхронизировать целый ряд сервисов, в которых будут автоматически публиковаться посты, а также появится ссылка на аккаунт. В данной статье мы разберемся, как сделать ссылку на Инстаграм в ВК несколькими способами.
Например, пользователи Instagram могут синхронизировать целый ряд сервисов, в которых будут автоматически публиковаться посты, а также появится ссылка на аккаунт. В данной статье мы разберемся, как сделать ссылку на Инстаграм в ВК несколькими способами.
Также предлагаем ознакомиться с небольшим роликом. Из него вы сможете узнать о всех подробностях и нюансах прикрепления аккаунта Instagram к VK.
Вставка ссылки в описание страницы ВК
Сначала рассмотрим стандартный функционал социальной сети ВКонтакте. Через редактирование личной страницы вы сможете привязать аккаунт Instagram, после чего он будет отображаться в описании профиля. Для этого необходимо сделать следующее:
- Зайти на сайт соцсети и авторизоваться.
- Перейти к разделу редактирования с помощью меню, которое открывается при нажатии на аватар в правой верхней части интерфейса.
- Теперь необходимо переключиться ко вкладке «Контакты».
- В нижней части настроек находится кнопка «Настроить импорт» возле иконки Инстаграма. Нажмите ее.
- После этого откроется дополнительное окно с формой авторизации в Instagram. Введите свои логин и пароль.
- Если данные введены правильно, то окно закроется, а вместо него на сайте ВК появятся параметры интеграции с соцсетью. Здесь вы можете привязать другой профиль, выбрать тип импорта фотографий, а также альбом для них. После настроек нажмите «Сохранить».
После выполненных действий вы можете проверить работоспособность метода. Для этого достаточно перейти на свою страницу и открыть блок «Контактные данные». Напротив строки Instagram будет стоять активная ссылка с вашим никнеймом.
Автоматическая публикация фото
Если вы настроите автоматическую отправку всех постов в ВК, то они будут появляться на стене страницы и сохраняться в специальный альбом с описанием из Instagram.
Мнение эксперта
Дарья Ступникова
Специалист по WEB-программированию и компьютерным системам. Редактор PHP/HTML/CSS сайта os-helper.ru.
Спросить у ДарьиЭтот функционал работает только на фотографии. Вы не сможете интегрировать Stories или видеоролики.
Привязку к аккаунту вы можете выполнить и через приложение Instagram. Для этого даже необязательно пользоваться смартфоном – вы можете установить программу через эмулятор или скачать официальную утилиту из Microsoft Store. Приложение на ПК полностью копирует интерфейс и функционал мобильной версии, поэтому представленную инструкцию можно считать универсальной:
- Сначала перейдите во вкладку со своим профилем.
- Откройте раздел настроек.
- Перейдите во вкладку «Связанные аккаунты».
- Здесь находится список социальных сетей, которые можно интегрировать с Инстаграмом. Выберите ВКонтакте.
- После этого появится окно VK, в котором нужно авторизоваться с помощью логина и пароля от соцсети.

После авторизации напротив названия социальной сети появится имя вашей страницы VK. Если еще раз нажать на этот пункт, то появится кнопка отмены интеграции двух аккаунтов. Когда вы уберете данную опцию, публикации перестанут дублироваться во ВКонтакте, несмотря на выставленные параметры в соцсети.
Копирование ссылки для использования в ВК
Напоследок рассмотрим ситуацию, когда вам не нужно интегрировать два сервиса, а необходимо просто вставить ссылку на Instagram в сообщение, пост на стену и так далее. Для этого пользователь должен скопировать адрес страницы или публикации через мобильное приложение или официальный сайт. В первом случае вам необходимо зайти в профиль человека и нажать на кнопку с тремя точками. После этого в меню выбрать пункт «Скопировать URL профиля».
Похожим образом работает и официальный сайт, только вам придется копировать адрес из поисковой строки вручную. При загрузке сайта Instagram не соглашайтесь на переход в приложение, а оставайтесь в браузере. После этого откройте нужный профиль и скопируйте адресную строку в буфер обмена.
Заключение
Итак, мы разобрались, какие действия позволяют делать ссылки на Инстаграм во ВКонтакте. Если вы хотите синхронизировать социальные сети, то обязательно привяжите аккаунт Instagram, а также добавьте ссылку на свой профиль в описание страницы ВК.
VK. Pyrus Help
Если клиенты обращаются к вам за помощью во ВКонтакте (vk.com), подключите аккаунт службы поддержки к форме обращения клиента в Pyrus.
1. Войдите раздел пользователя в нижнем левом углу экрана. Выберите вкладку Расширения, а затем на странице доступных интеграций нажмите иконку
Выберите вкладку Расширения, а затем на странице доступных интеграций нажмите иконку
Важно! Для доступа в раздел Расширения из профиля пользователя вам необходимы права Менеджера.
2.Выберите форму Pyrus, с которой будет интегрирована ваша группа ВКонтакте.
Также вы можете подключить ВКонтакте в настройках нужной формы во вкладке Расширения.
3. Теперь нужно связать информацию из сообщения или комментария к посту в VK и поля выбранной формы. Это можно сделать в один клик, нажав на ссылку Создать недостающие поля.
Готово! В заявке из комментария будут отображаться имя отправителя, само сообщение, ссылка на профиль пользователя в ВКонтакте и ссылка на пост, под которым оставлен комментарий.
4. Далее следует решить, как вы будете получать повторные комментарии от клиента в ветке обсуждения к посту или в сообщениях — в виде новой заявки или в виде комментария к существующей. Сделайте выбор и нажмите Сохранить.
Сделайте выбор и нажмите Сохранить.
5. Пришло время привязать к форме страницу вашей группы в VK. Выберите из списка или создайте нужную группу.
6. Введите ключ доступа к вашей группе ВКонтакте (API-токен). Чтобы получить его, войдите в аккаунт группы и в разделе
Чтобы скопировать ключ, нажмите Показать и следуйте подсказкам сервиса.
Если у вас нет API-ключа, нажмите Создать и следуйте дальнейшим подсказкам.
7. Убедитесь, что в вашей группе включены сообщения. Для этого перейдите в управление группы, выберите Сообщения и активируйте режим Включены.
8. Чтобы поддерживать функции бота, такие как запрос оценки сервиса, откройте Настройки бота, включите их и сохраните изменения.
9. Активируйте расширение.
Сообщения и комментарии, которые пользователи отправляют в вашу группу во ВКонтакте, будут автоматически появляться во Входящих у специалистов поддержки в Pyrus. Как и с другими каналами, в заявке будет отдельная вкладка для переписки с клиентом и отдельная вкладка — для переписки с коллегами в Pyrus. Ваши ответные сообщения клиент будет получать у себя во ВКонтакте.
Создание новой заявки из старой переписки
Иногда клиенты пишут в старую заявку по другой теме. Например, человек обращался, потому что не получалось оплатить заказ, а спустя неделю решил уточнить условия обслуживания и написал в ответ на старую переписку со службой поддержки. Но тема новая, и для статистики лучше, чтобы это была отдельная заявка.
Зайдите в настройку формы и в блоке Расширения и выберите ВКонтакте. На открывшейся странице внизу найдите раздел Расширенные настройки. Поставьте галку и задайте, через сколько дней или часов после закрытия заявки повторные обращения клиента будут становиться новыми заявками. Нажмите Сохранить.
Нажмите Сохранить.
Автоматическое заполнение полей заявки
Если обращениями из ВКонтакте занимается отдельная группа поддержки, удобно автоматически отмечать источник заявки. Для этого настройте автозаполнение поля
Откройте настройку формы. В Расширениях нажмите в поле ВКонтакте.
В блоке Автоматически изменить поля нажмите Добавить поле и установите для поля Тип заявки вариант ВКонтакте.
Так же вы можете настроить автозаполнение любого поля типа Выбор и Справочник в форме заявки.
Как связать или отвязать Инстаграм с Вконтакте: инструкция пользователя
Автор Лика Торопова Опубликовано Обновлено
Соединение между разными социальными сетями поможет в восстановлении доступа, передаче личных данных и импортировании изображений. Есть несколько способов, как связать Инстаграм с ВК.
Связываем Instagram с VK
Мнгоие сталкивались с такой проблемой синхронизации Инстаграма с Контактом. И делается это вполне просто.
Инструкция, как связать два аккаунта:
- Открыть свою страницу в ВК.
- Перейти в «Редактирование» – Контакты.Затем в списке опций нужно найти «Связанные аккаунты».
- Появится раздел «Импорт с Инстаграмом» – ввести данные от социальной сети.
Данная операция поможет связать Инстаграм и Контакт на компьютерной версии. Все записи пользователя автоматически импортируются Vkontakte, оставляя снизу подпись «repost Instagram» и ссылку на учетную запись.
Как отвязать Инстаграм от ВК
Если появилась необходимость отвязать Инстаграм от ВК, то следует выполнить следующие действия:
- Для начала необходимо зайти в ВК.
- Перейти в «Настройки»– «Контакты».
- Если ВК привязан к Инстаграму, то он будет синим цветом.
- Далее следует нажать по надписи «Вконтакте» и нажать «Отменить связь».
Данный алгоритм действия поможет отвязать Инстаграм от Контакта.
Связь между группой VK и Instagram
На данный момент связь группы ВК и Инстаграма очень сложна. В данной ситуации на помощь приходит специальный сайт «onemorepost.ru».
Вам необходимо:
- Открыть сайт и пройти регистрацию – авторизоваться.
- Необходимо зайти на панель управления, где находится вкладка аккаунтов.
- Добавляем аккаунт группы в ВК и Инстаграм.
- Подтвердите права приложения – Создать аккаунт связи.
Сервис платный, но доступна одна бесплатная проверка. Также, можно оставить ссылку на публикацию, чтобы отправить одно фото или видео.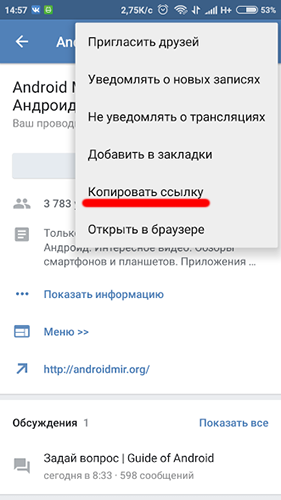
Мнение эксперта
Лика Торопова
Много времени провожу в социальных сетях, любимчик — Instagram
Привет, я автор сайта storins.ru, если вы не нашли своего ответа, или у вас есть вопросы, то напишите мне, я постараюсь ответить вам максимально быстро и решить вашу проблему. Список самых интересных моих статей:Используем ссылку на публикацию:
- Открыть нужный пост в Instagram в браузере и скопировать ссылку из адресной строки.
- Открыть VK – заполнить описание.
- В запись вставить ссылку.
- После того как появилась фото, которое используется в посте, ссылку можно удалить. Вместо нее можно написать короткую информацию о содержании поста.
Пользователю доступен только ручной способ ввода ссылок и обмена медиафайлами между социальными сетями. Сделать прямое подключение не получится из-за проблем авторизации с профилем ВК в Инстаграме.
Репост ссылок из ВК в Инстаграм и наоборот
Отправить ссылку из публикации ВК в Инстаграм можно только через копирование. Пользователь должен перейти в свой профиль Вконтакте и скопировать нужный адрес. При этом, в Instagram не появится активная ссылка: как и другие, она будет некликабельна.
Пользователь должен перейти в свой профиль Вконтакте и скопировать нужный адрес. При этом, в Instagram не появится активная ссылка: как и другие, она будет некликабельна.
Чтобы скопировать ссылку из ВК и вставить в Инстаграм: нужно перейти в первую социальную сеть и нажать на нужную публикацию. После – вернуться во вторую учетную запись и просто вставить ссылку. При этом, посетители страницы не смогут просто перейти по указанному адресу. Его нужно снова копировать и вставлять в адресную строку.
Проблемы с импортом данных из Вконтакте в Инстаграм и наоборот, возникают только со стороны фото-сети. То есть, подключить страницу из ВК к Instagram можно, а вот в обратную сторону – нельзя.
С проблемой пользователи столкнулись год назад, когда связь между профилями была утеряна. На данный момент, Facebook не делает никаких заявлений по поводу решения ошибки авторизации.
Как получить прямую ссылку на товар или категорию – Центр поддержки
Если вы хотите кому-то отправить прямую ссылку на товар или категорию, вы легко можете скопировать ссылку из витрины вашего магазина. Убедитесь, что товар или категория, на которую вы хотите получить ссылку, включен:
Убедитесь, что товар или категория, на которую вы хотите получить ссылку, включен:
Как получить прямую ссылку на товар или категорию
Чтобы получить прямую ссылку на товар или категорию:
- Откройте витрину магазина.
- Откройте товар, на который вы хотите получить ссылку, или категорию.
- Скопируйте ссылку из адресной строки браузера:
Готово! Вы скопировали прямую ссылку на товар или категорию в вашем магазине.
Как получить прямую ссылку на скрытый товар или категорию
Если в вашем магазине есть скрытые товары или категории, вы также можете получить на них ссылку. Если вы отправите эту ссылку покупателю, они смогут открыть товар или категорию.
Ссылка на скрытый товар
Чтобы получить ссылку на скрытый товар, перейдите в панели управления Эквида на страницу Каталог → Товары. Откройте товар, на который вы хотите получить ссылку, и нажмите Открыть на витрине. Вам откроется страница товара на витрине:
Ссылка на скрытую категорию
Чтобы получить ссылку на скрытую категорию, вы можете временно включить категорию в магазине, открыть витрину, открыть страницу категории и скопировать ссылку из адресной строки браузера. После этого снова скройте категорию.
После этого снова скройте категорию.
Другой вариант — сформировать ссылку на скрытую категорию, используя адрес магазина и внутренний ID категории:
- В панели управления Эквида перейдите в Каталог → Категории.
- Нажмите на скрытую категорию.
- В адресной строке браузера скопируйте ID категории. Это комбинация цифр, которая идёт после category:id=:
- Подставьте ID категории после адреса своего магазина и добавьте букву c. У вас получится ссылка вида: www.example.com/-c12345, где 12345 — это ID категории.
Обязательно откройте полученную ссылку в браузере, чтобы убедиться, что она работает и открывает нужную вам категорию. После этого вы можете делиться ссылкой.
Была ли эта статья полезной?
Приятно! Спасибо за ваш отзыв.
Спасибо за ваш отзыв.
Простите за это! Что было не так?
Отправить отзыв Пользователи, считающие этот материал полезным: 81 из 244jef.vk, Автор на ObsEva — Страница 5 из 7
- Подпишитесь на новости Опубликовано автор jef.vk в Подпишитесь на новости
- Профиль компании Опубликовано автор: jef.vk в профиль компании и блок страницы
- Форма партнерства Опубликовано автор: jef.vk в Блок страниц и партнерство
- Изображение посадки партнерства Размещено автором jef.vk в Блок страниц и партнерство
- Преждевременные роды Опубликовано автор jef.vk в OBE022 и блок страницы
- Изображение посадки Nolasiban Размещено автором jef.vk в OBE022 и блок страницы
- Вспомогательные репродуктивные технологии (ВРТ) Опубликовано автором jef.vk в Ноласибан и Пейдж блок
- Изображение посадки ноласибана Размещено автором jef.vk в Ноласибан и Пейдж блок
- Миома матки Опубликовано автором jef.vk в Linzagolix и Пейдж блок
- Эндометриоз Опубликовано автор jef.vk в Линзаголикс и Пейдж блок
Вы уверены, что хотите покинуть ObsEva.com?
Сожалеем, что вы уходите.Выберите одно из следующих действий, чтобы остаться на сайте или уйти.
Оставайтесь на ObsEva.com Продолжить ссылкуЗаявление об ограничении ответственности
Обратите внимание, что следующая информация содержит информацию об исследуемых лекарственных средствах. Эти продукты еще не одобрены для продажи Европейским агентством по лекарственным средствам или Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США и все еще находятся в стадии разработки. Для выдачи разрешения на продажу могут потребоваться дополнительные дополнительные исследования, которые могут зависеть от множества факторов и не гарантируются. Нажмите «вернуть» , если вы не желаете получать такую информацию. Нажимая «продолжить» , вы подтверждаете, что хотите получать научную информацию о наших исследуемых продуктах.
Эти продукты еще не одобрены для продажи Европейским агентством по лекарственным средствам или Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США и все еще находятся в стадии разработки. Для выдачи разрешения на продажу могут потребоваться дополнительные дополнительные исследования, которые могут зависеть от множества факторов и не гарантируются. Нажмите «вернуть» , если вы не желаете получать такую информацию. Нажимая «продолжить» , вы подтверждаете, что хотите получать научную информацию о наших исследуемых продуктах.
Заявление об ограничении ответственности
Обратите внимание, что следующая информация содержит информацию об исследуемых лекарственных средствах. Эти продукты еще не одобрены для продажи Европейским агентством по лекарственным средствам или США.S. Управление по контролю за продуктами и лекарствами и все еще находятся в стадии разработки. Для выдачи разрешения на продажу могут потребоваться дополнительные дополнительные исследования, которые могут зависеть от множества факторов и не гарантируются.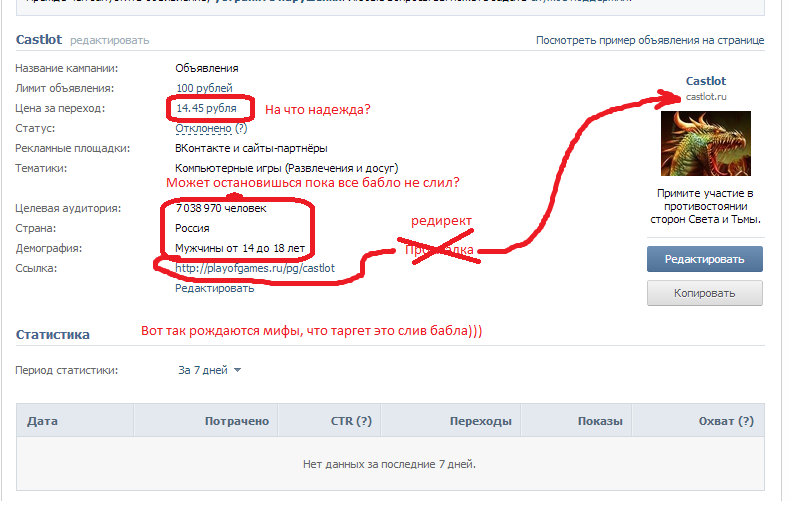 Нажмите «вернуть» , если вы не желаете получать такую информацию. Нажимая «продолжить» , вы подтверждаете, что хотите получать научную информацию о наших исследуемых продуктах.
Нажмите «вернуть» , если вы не желаете получать такую информацию. Нажимая «продолжить» , вы подтверждаете, что хотите получать научную информацию о наших исследуемых продуктах.
Анализ обогащения набора генов: основанный на знаниях подход к интерпретации полногеномных профилей экспрессии
Реферат
Хотя анализ экспрессии РНК в масштабе всего генома стал обычным инструментом в биомедицинских исследованиях, извлечение биологической информации из такой информации остается серьезной проблемой.Здесь мы описываем мощный аналитический метод, называемый анализом обогащения генетического набора (GSEA), для интерпретации данных экспрессии генов. Эффективность этого метода достигается за счет сосредоточения внимания на наборах генов, то есть на группах генов, которые имеют общую биологическую функцию, хромосомное положение или регуляцию. Мы демонстрируем, как GSEA помогает понять несколько наборов данных, связанных с раком, включая лейкемию и рак легких. Примечательно, что там, где анализ одного гена обнаруживает небольшое сходство между двумя независимыми исследованиями выживаемости пациентов с раком легких, GSEA выявляет много общих биологических путей.Метод GSEA воплощен в свободно доступном программном пакете вместе с исходной базой данных из 1325 биологически определенных наборов генов.
Примечательно, что там, где анализ одного гена обнаруживает небольшое сходство между двумя независимыми исследованиями выживаемости пациентов с раком легких, GSEA выявляет много общих биологических путей.Метод GSEA воплощен в свободно доступном программном пакете вместе с исходной базой данных из 1325 биологически определенных наборов генов.
Анализ экспрессии в масштабе всего генома с помощью ДНК-микрочипов стал основой геномных исследований ( 1, 2). Проблема больше не в получении профилей экспрессии генов, а в интерпретации результатов для понимания биологических механизмов.
В типичном эксперименте профили экспрессии мРНК генерируются для тысяч генов из коллекции образцов, принадлежащих к одному из двух классов, например, опухоли, которые чувствительны против опухолей.устойчив к препарату. Гены можно упорядочить в ранжированном списке L в соответствии с их дифференциальной экспрессией между классами. Задача состоит в том, чтобы извлечь смысл из этого списка.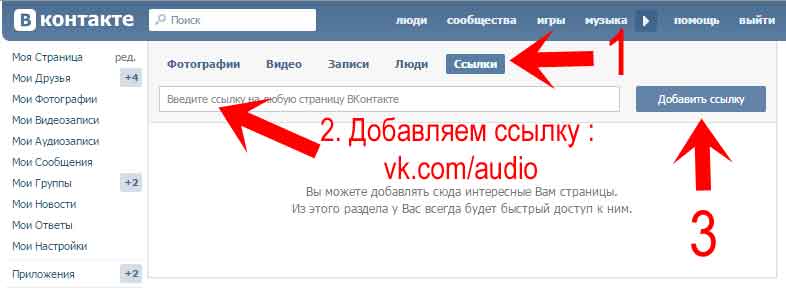
Обычный подход предполагает сосредоточение внимания на нескольких генах вверху и внизу L (т.е. тех, которые демонстрируют наибольшее различие), чтобы выявить контрольные биологические ключи. У этого подхода есть несколько основных ограничений.
( i ) После корректировки для проверки нескольких гипотез ни один отдельный ген не может соответствовать порогу статистической значимости, потому что соответствующие биологические различия незначительны по сравнению с шумом, присущим технологии микрочипов.
( ii ) В качестве альтернативы можно оставить длинный список статистически значимых генов без какой-либо объединяющей биологической темы. Интерпретация может быть сложной и произвольной, поскольку зависит от области знаний биолога.
( iii ) Анализ одного гена может упустить важные эффекты на пути. Клеточные процессы часто влияют на совокупность генов, действующих согласованно. Повышение на 20% всех генов, кодирующих элементы метаболического пути, может резко изменить поток через этот путь и может быть более важным, чем 20-кратное увеличение одного гена.
( iv ) Когда разные группы изучают одну и ту же биологическую систему, список статистически значимых генов из двух исследований может показывать удручающе небольшое совпадение ( 3).
Чтобы преодолеть эти аналитические проблемы, мы недавно разработали метод, называемый анализом обогащения набора генов (GSEA), который оценивает данные микрочипов на уровне наборов генов. Наборы генов определяются на основе предшествующих биологических знаний, например, опубликованной информации о биохимических путях или коэкспрессии в предыдущих экспериментах.Цель GSEA — определить, имеют ли члены набора генов S тенденцию встречаться в верхней (или нижней) части списка L , и в этом случае набор генов коррелирует с различием фенотипического класса.
Мы использовали предварительную версию GSEA для анализа данных биопсии мышц диабетиков и здоровых людей в контрольной группе ( 4). Метод показал, что гены, участвующие в окислительном фосфорилировании, демонстрируют пониженную экспрессию у диабетиков, хотя среднее снижение на ген составляет всего 20%. Результаты этого исследования были независимо подтверждены другими исследованиями микрочипов ( 5) и in vivo функциональных исследований ( 6).
Результаты этого исследования были независимо подтверждены другими исследованиями микрочипов ( 5) и in vivo функциональных исследований ( 6).
Учитывая этот успех, мы разработали GSEA в виде надежного метода анализа данных молекулярного профилирования. Мы изучили его характеристики и производительность, а также существенно переработали и обобщили оригинальный метод для более широкого применения.
В этой статье мы даем полное математическое описание методологии GSEA и демонстрируем ее полезность, применяя ее к нескольким разнообразным биологическим проблемам.Мы также создали программный пакет под названием gsea-p и начальный перечень наборов генов (Molecular Signature Database, MSigDB), оба из которых находятся в свободном доступе.
Методы
Обзор GSEA. GSEA рассматривает эксперименты с полногеномными профилями экспрессии из образцов, принадлежащих к двум классам, обозначенным 1 или 2. Гены ранжируются на основе корреляции между их экспрессией и различием классов с использованием любой подходящей метрики ( Рис.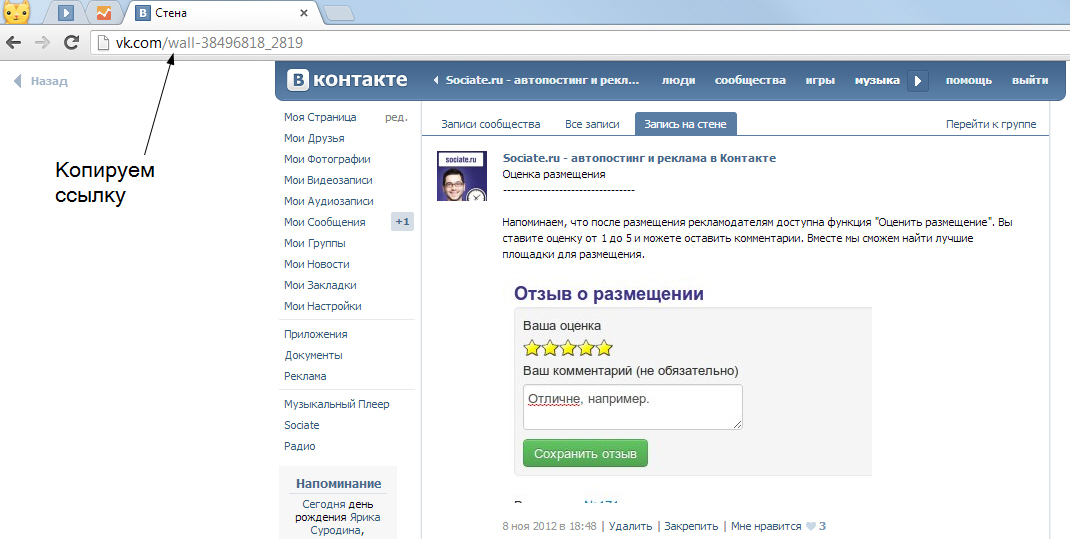 1 A ).
1 A ).
Обзор GSEA, иллюстрирующий метод. ( A ) Набор данных экспрессии, отсортированный по корреляции с фенотипом, соответствующей тепловой картой и «тегами генов», то есть местоположением генов из набора S в отсортированном списке. ( B ) График текущей суммы для S в наборе данных, включая расположение максимальной оценки обогащения ( ES ) и переднего подмножества.
Учитывая априори определенный набор генов S (e.g., гены, кодирующие продукты метаболического пути, расположенные в одной и той же цитогенетической полосе или имеющие одну и ту же категорию GO), цель GSEA состоит в том, чтобы определить, распределены ли члены S случайным образом по L или первоначально обнаружены вверху или внизу. Мы ожидаем, что наборы, связанные с фенотипическими различиями, будут иметь тенденцию к последнему распределению.
Есть три ключевых элемента метода GSEA:
Шаг 1: Расчет оценки обогащения.
Мы вычисляем оценку обогащения ( ES ), которая отражает степень, в которой набор S перепредставлен в крайних точках (вверху или внизу) всего ранжированного списка L . Оценка рассчитывается путем перехода по списку L , увеличения статистики промежуточной суммы, когда мы встречаем ген в S и уменьшения ее, когда мы встречаем гены не в S. Величина приращения зависит от корреляции гена с фенотипом.Оценка обогащения — это максимальное отклонение от нуля, встречающееся при случайном блуждании; он соответствует взвешенной статистике типа Колмогорова – Смирнова (см. 7 и Рис.1 B ).Шаг 2: Оценка уровня значимости ES. Мы оцениваем статистическую значимость (номинальное значение P ) ES с помощью эмпирической процедуры перестановочного теста на основе фенотипа, которая сохраняет сложную корреляционную структуру данных экспрессии генов.В частности, мы переставляем метки фенотипа и повторно вычисляем ES набора генов для переставленных данных, что генерирует нулевое распределение для ES.
Затем вычисляется эмпирическое номинальное значение P наблюдаемого ES относительно этого нулевого распределения. Важно отметить, что перестановка меток классов сохраняет корреляции ген-ген и, таким образом, обеспечивает более биологически обоснованную оценку значимости, чем было бы получено путем перестановки генов.Шаг 3: Корректировка для проверки множественных гипотез. Когда оценивается вся база данных наборов генов, мы корректируем предполагаемый уровень значимости, чтобы учесть проверку нескольких гипотез. Сначала мы нормализуем ES и для каждого набора генов, чтобы учесть размер набора, давая нормализованный показатель обогащения ( NES ). Затем мы контролируем долю ложных срабатываний, вычисляя коэффициент ложного обнаружения (FDR) ( 8, 9) соответствует каждому NES .FDR — это оценочная вероятность того, что набор с данным NES представляет ложноположительный результат; он вычисляется путем сравнения хвостов наблюдаемого и нулевого распределений для NES .
 Мы вычисляем оценку обогащения ( ES ), которая отражает степень, в которой набор S перепредставлен в крайних точках (вверху или внизу) всего ранжированного списка L . Оценка рассчитывается путем перехода по списку L , увеличения статистики промежуточной суммы, когда мы встречаем ген в S и уменьшения ее, когда мы встречаем гены не в S. Величина приращения зависит от корреляции гена с фенотипом.Оценка обогащения — это максимальное отклонение от нуля, встречающееся при случайном блуждании; он соответствует взвешенной статистике типа Колмогорова – Смирнова (см. 7 и Рис.1 B ).
Мы вычисляем оценку обогащения ( ES ), которая отражает степень, в которой набор S перепредставлен в крайних точках (вверху или внизу) всего ранжированного списка L . Оценка рассчитывается путем перехода по списку L , увеличения статистики промежуточной суммы, когда мы встречаем ген в S и уменьшения ее, когда мы встречаем гены не в S. Величина приращения зависит от корреляции гена с фенотипом.Оценка обогащения — это максимальное отклонение от нуля, встречающееся при случайном блуждании; он соответствует взвешенной статистике типа Колмогорова – Смирнова (см. 7 и Рис.1 B ). Затем вычисляется эмпирическое номинальное значение P наблюдаемого ES относительно этого нулевого распределения. Важно отметить, что перестановка меток классов сохраняет корреляции ген-ген и, таким образом, обеспечивает более биологически обоснованную оценку значимости, чем было бы получено путем перестановки генов.
Затем вычисляется эмпирическое номинальное значение P наблюдаемого ES относительно этого нулевого распределения. Важно отметить, что перестановка меток классов сохраняет корреляции ген-ген и, таким образом, обеспечивает более биологически обоснованную оценку значимости, чем было бы получено путем перестановки генов.
Детали реализации описаны в Приложении (см. Также Вспомогательный текст , который опубликован в качестве вспомогательной информации на веб-сайте PNAS).
Отметим, что метод GSEA по нескольким важным отличиям от предварительной версии (см. Вспомогательный текст ).В исходной реализации статистика промежуточной суммы использовала одинаковые веса на каждом шаге, что давало высокие баллы для наборов, сгруппированных около середины ранжированного списка ( Рис.2 и Таблица 1). Эти наборы не представляют биологически значимой корреляции с фенотипом. Мы решили эту проблему, взвешивая шаги в соответствии с корреляцией каждого гена с фенотипом. Мы заметили, что использование взвешенных шагов может привести к тому, что распределение наблюдаемых баллов ES будет асимметричным в тех случаях, когда гораздо больше генов коррелируют с одним из двух фенотипов.Поэтому мы оцениваем уровни значимости, рассматривая отдельно наборы генов с положительной и отрицательной оценкой (, приложение ; см. Также рис. 4, который опубликован в качестве вспомогательной информации на веб-сайте PNAS).
Также рис. 4, который опубликован в качестве вспомогательной информации на веб-сайте PNAS).
Оригинал ( 4) поведение по счету обогащения. Распределение трех наборов генов из функциональной коллекции C2 в списке генов в примере линии мужских / женских лимфобластоидных клеток, ранжированных по их корреляции с полом: S1, набор генов инактивации хромосомы X; S2 — путь, описывающий импорт витамина С в нейроны; S3, относящийся к хемокиновым рецепторам, экспрессируемым Т-хелперами.Показаны графики текущей суммы для трех наборов генов: S1 значительно обогащен самками, как и ожидалось, S2 распределен случайным образом и имеет низкую оценку, а S3 не обогащен в верхней части списка, но не является случайным, поэтому он имеет хорошие оценки. Стрелки показывают положение максимальной оценки обогащения и точку, где корреляция (отношение сигнал / шум) пересекает ноль. В таблице 1 сравниваются номинальные значения P для S1, S2 и S3 с использованием исходного и нового методов. Новый метод снижает значимость таких наборов, как S3.
Новый метод снижает значимость таких наборов, как S3.
Наша предварительная реализация использовала другой подход, частоту ошибок по семействам (FWER), для исправления нескольких проверок гипотез. FWER — это консервативная коррекция, которая направлена на то, чтобы список сообщаемых результатов не включал даже один ложноположительный набор генов. Этот критерий оказался настолько консервативным, что многие приложения не дали статистически значимых результатов.Поскольку наша основная цель — генерировать гипотезы, мы решили использовать FDR, чтобы сосредоточиться на контроле вероятности того, что каждый отчетный результат является ложноположительным.
Основываясь на нашем статистическом анализе и эмпирической оценке, GSEA показывает широкую применимость. Он может обнаруживать тонкие сигналы обогащения и сохраняет наши исходные результаты в исх. 4, причем путь окислительного фосфорилирования значительно обогащен в нормальных образцах ( P = 0,008, FDR = 0,04).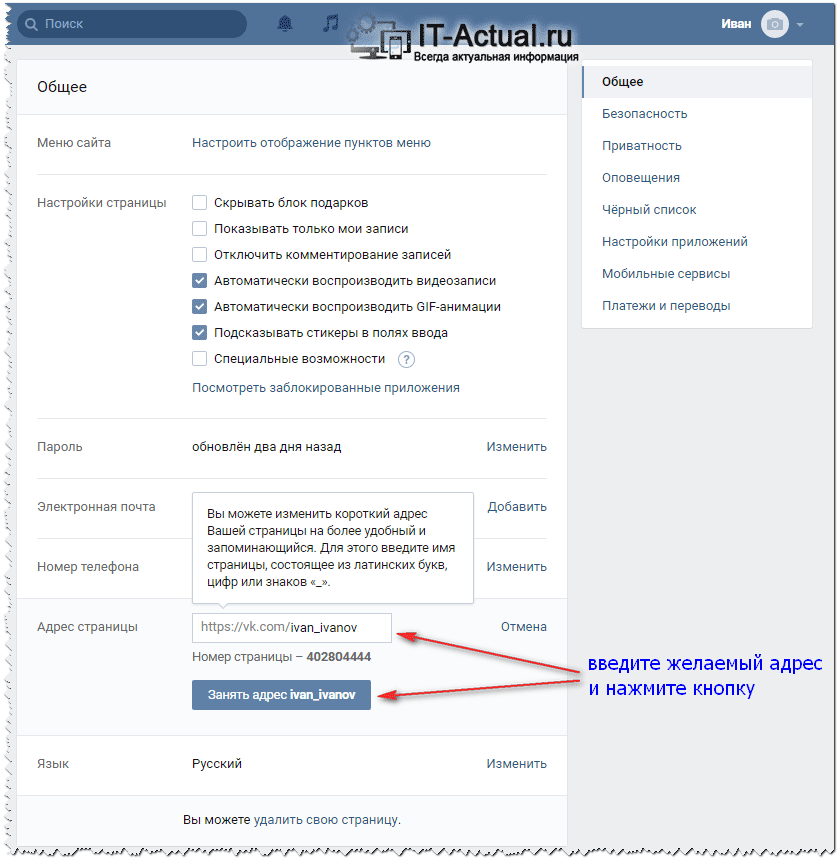 Эта методология была реализована в программном средстве под названием gsea-p.
Эта методология была реализована в программном средстве под названием gsea-p.
Передовое подмножество. Наборы генов можно определить с помощью множества методов, но не все члены набора генов обычно будут участвовать в биологическом процессе. Часто бывает полезно выделить основные члены наборов генов с высокими показателями, которые вносят вклад в ES . Мы определяем передовое подмножество как те гены в наборе генов S , которые появляются в ранжированном списке L в точке или раньше точки, в которой текущая сумма достигает максимального отклонения от нуля ( Инжир.1 В ). Подмножество переднего края можно интерпретировать как ядро набора генов, который отвечает за сигнал обогащения.
Исследование передового подмножества может выявить биологически важное подмножество в наборе генов, как мы покажем ниже в нашем анализе статуса P53 в линиях раковых клеток. Этот подход особенно полезен с вручную подобранными наборами генов, которые могут представлять собой объединение взаимодействующих процессов. Мы впервые наблюдали этот эффект в нашем предыдущем исследовании ( 4), где мы вручную определили два набора с высокими оценками, курируемый путь и кластер, полученный с помощью вычислений, которые разделяют большое подмножество генов, позже подтвержденных как ключевой регулон, измененный при диабете человека.
Мы впервые наблюдали этот эффект в нашем предыдущем исследовании ( 4), где мы вручную определили два набора с высокими оценками, курируемый путь и кластер, полученный с помощью вычислений, которые разделяют большое подмножество генов, позже подтвержденных как ключевой регулон, измененный при диабете человека.
Наборы генов с высокими показателями можно сгруппировать на основе передовых подмножеств генов, которые они разделяют. Такое группирование может показать, какие из этих наборов генов соответствуют одним и тем же биологическим процессам, а какие представляют различные процессы.
Программный пакет gsea-p включает инструменты для изучения и кластеризации передовых подмножеств ( Supporting Text ).
Варианты метода GSEA. Мы сосредотачиваемся выше и в результатах на использовании GSEA для анализа ранжированного списка генов, отражающего дифференциальную экспрессию между двумя классами, каждый из которых представлен большим количеством образцов. Однако этот метод можно применить к ранжированным спискам генов, возникающим в других условиях.
Однако этот метод можно применить к ранжированным спискам генов, возникающим в других условиях.
Гены могут быть ранжированы на основе различий, наблюдаемых в небольшом наборе данных, при слишком малом количестве выборок, чтобы обеспечить строгую оценку уровней значимости путем перестановки меток классов. В этих случаях значение P можно оценить путем перестановки генов, в результате чего гены случайным образом распределяются по наборам при сохранении их размера. Этот подход не совсем точен: поскольку он игнорирует корреляции ген-ген, он будет переоценивать уровни значимости и может привести к ложноположительным результатам.Тем не менее, это может быть полезно для генерации гипотез. Программа gsea-p поддерживает эту опцию.
Гены также можно ранжировать на основе того, насколько хорошо их экспрессия коррелирует с заданным целевым паттерном (например, паттерном экспрессии конкретного гена). В Lamb et al. ( 10), процедура, подобная GSEA, использовалась для демонстрации обогащения набора мишеней списка циклина D1, ранжированного по корреляции с профилем циклина D1 в компендиуме типов опухолей. Опять же, приблизительные значения P можно оценить путем перестановки генов.
Опять же, приблизительные значения P можно оценить путем перестановки генов.
Первоначальный каталог наборов генов человека. GSEA оценивает набор данных микрочипа запроса, используя набор наборов генов. Поэтому мы создали первоначальный каталог из 1325 наборов генов, который мы назвали MSigDB 1.0 ( Supporting Text ; см. Также Таблицу 3, которая опубликована в качестве вспомогательной информации на веб-сайте PNAS), состоящий из четырех типов наборов.
Цитогенетические наборы (C 1 , 319 наборов генов). Этот каталог включает 24 набора, по одному для каждой из 24 хромосом человека, и 295 наборов, соответствующих цитогенетическим полосам.Эти наборы помогают идентифицировать эффекты, связанные с хромосомными делециями или амплификациями, дозовой компенсацией, эпигенетическим молчанием и другими региональными эффектами.
Функциональные наборы (C 2 , 522 генных набора). Этот каталог включает 472 набора, содержащих гены, продукты которых участвуют в определенных метаболических и сигнальных путях, о чем сообщается в восьми общедоступных, вручную отобранных базах данных, и 50 наборов, содержащих гены, координируемые в ответ на генетические и химические нарушения, как сообщается в различных экспериментальных работах. .
.
Наборы регуляторных мотивов (C 3 , 57 наборов генов). Этот каталог основан на нашей недавней работе, в которой сообщается о 57 обычно консервативных регуляторных мотивах в промоторных областях генов человека ( 11) и позволяет связать изменения в эксперименте с микрочипами с консервативным предполагаемым цис-регуляторным элементом.
Наборы соседей (C 4 , 427 наборов генов). Эти наборы определяются соседями экспрессии, сосредоточенными на генах, связанных с раком.
Эта база данных предоставляет начальную коллекцию наборов генов для использования с GSEA и иллюстрирует типы наборов генов, которые могут быть определены, в том числе основанные на предварительных знаниях или полученные путем вычислений.
Программное обеспечение gsea-p и наборы генов MSigDB. Чтобы облегчить использование GSEA, мы разработали ресурсы, которые можно бесплатно получить в Broad Institute по запросу. Эти ресурсы включают программное обеспечение gsea-p, MSigDB 1.0 и сопроводительную документацию.
Эти ресурсы включают программное обеспечение gsea-p, MSigDB 1.0 и сопроводительную документацию.
Программное обеспечение доступно как ( i ) платформо-независимое настольное приложение с графическим пользовательским интерфейсом; ( ii ) программы на языках r и java, которые опытные пользователи могут включать в свои собственные анализы или программные среды; ( iii ) аналитический модуль в нашем пакете анализа микрочипов генов (доступен по запросу) ( iv ) будущий веб-сервер GSEA, позволяющий пользователям проводить собственный анализ непосредственно на веб-сайте.Подробный пример формата вывода GSEA доступен на сайте, а также в Supporting Text .
Результаты
Мы исследовали способность GSEA предоставлять биологически значимые идеи на шести примерах, по которым имеется значительная справочная информация. В каждом случае мы искали значительно ассоциированные наборы генов из одного или обоих подкаталогов C1 и C2 (см. Выше).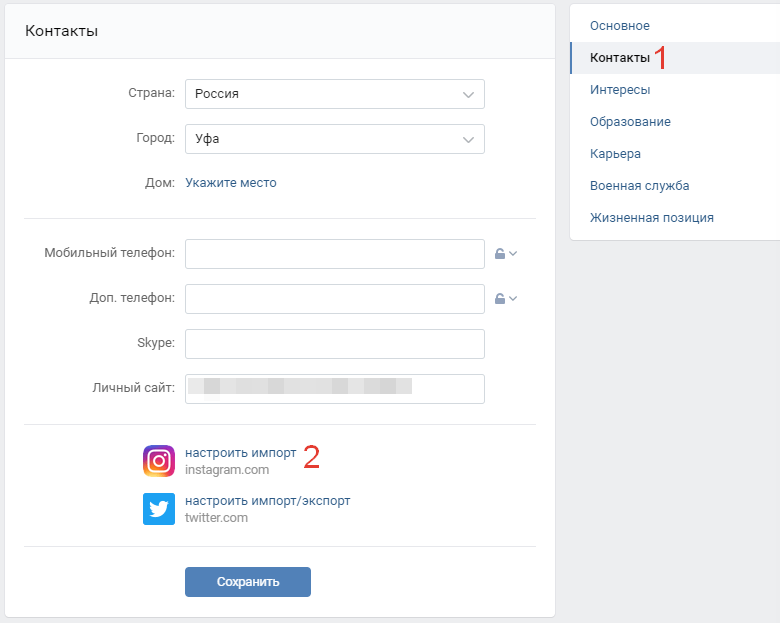 В таблице 2 перечислены все наборы генов с FDR ≤ 0,25.
В таблице 2 перечислены все наборы генов с FDR ≤ 0,25.
Мужские и женские лимфобластоидные клетки. В качестве простого теста мы сгенерировали профили экспрессии мРНК из линий лимфобластоидных клеток, полученных от 15 мужчин и 17 женщин (неопубликованные данные), и попытались идентифицировать наборы генов, коррелирующие с различиями «мужчина> женщина» и «женщина> мужчина».
Мы сначала протестировали обогащение наборов цитогенетических генов (C 1 ). Для сравнения мужчин и женщин мы ожидаем найти наборы генов на хромосоме Y. Действительно, GSEA продуцировал хромосому Y и две полосы Y, по крайней мере, с 15 генами (Yp11 и Yq11).Для сравнения самка> самец мы не ожидаем увидеть обогащение полос на хромосоме X, потому что большинство генов, сцепленных с X, подлежат дозовой компенсации и, следовательно, не более выражены у самок ( 12).
Далее мы рассмотрели обогащение функциональных наборов генов (C 2 ). В результате анализа получены три биологически информативных набора. Один состоит из генов, избегающих инактивации X [слитых из двух источников ( 13, 14), которые в значительной степени перекрываются], обнаружив ожидаемое обогащение женских клеток.Два дополнительных набора состоят из генов, обогащенных репродуктивными тканями (яичками и маткой), что примечательно тем, что экспрессия мРНК измерялась в лимфобластоидных клетках. Этот результат не просто связан с дифференциальной экспрессией генов на хромосомах X и Y, но остается значимым при ограничении аутосомными генами в наборах (Таблица 5, которая опубликована в качестве вспомогательной информации на веб-сайте PNAS).
В результате анализа получены три биологически информативных набора. Один состоит из генов, избегающих инактивации X [слитых из двух источников ( 13, 14), которые в значительной степени перекрываются], обнаружив ожидаемое обогащение женских клеток.Два дополнительных набора состоят из генов, обогащенных репродуктивными тканями (яичками и маткой), что примечательно тем, что экспрессия мРНК измерялась в лимфобластоидных клетках. Этот результат не просто связан с дифференциальной экспрессией генов на хромосомах X и Y, но остается значимым при ограничении аутосомными генами в наборах (Таблица 5, которая опубликована в качестве вспомогательной информации на веб-сайте PNAS).
Статус p53 в линиях раковых клеток. Затем мы исследовали паттерны экспрессии генов из коллекции линий раковых клеток NCI-60.Мы стремились использовать эти данные для идентификации мишеней фактора транскрипции p53, который регулирует экспрессию генов в ответ на различные сигналы клеточного стресса. Сообщалось о мутационном статусе гена p53 для 50 клеточных линий NCI-60, из которых 17 классифицированы как нормальные, а 33 как несущие мутации в гене ( 15).
Сначала мы применили GSEA для идентификации функциональных наборов генов (C 2 ), коррелирующих со статусом p53. Анализ p53 + > p53 — выявил пять наборов, экспрессия которых коррелирует с нормальной функцией p53 ( Таблица 2).Все они явно связаны с функцией p53. Наборы представляют собой (–) биологически аннотированный набор генов, кодирующих белки в пути передачи сигналов р53, который вызывает остановку клеточного цикла в ответ на повреждение ДНК; ( ii ) набор нижестоящих мишеней р53, определенных экспериментальной индукцией термочувствительного аллеля р53 в клеточной линии рака легкого; ( iii ) аннотированный набор индуцированных радиацией генов, ответ на которые, как известно, включает р53; ( iv ) аннотированный набор генов, индуцированных гипоксией, которые, как известно, действуют посредством р53-опосредованного пути, отличного от пути ответа на повреждение ДНК; и ( v ) аннотированный набор генов, кодирующих сигнальные пути белков теплового шока, которые защищают клетки от гибели в ответ на различные клеточные стрессы.
Дополнительный анализ (p53 — > p53 + ) идентифицирует один значимый набор генов: гены, участвующие в пути передачи сигналов Ras. Интересно, что два дополнительных набора, которые не достигают порога значимости, содержат гены, участвующие в сигнальных путях Ngf и Igf1. Чтобы выяснить, отражают ли эти три набора общую биологическую функцию, мы исследовали передовое подмножество для каждого набора генов (определенного выше). Передовые подмножества состоят из 16, 11 и 13 генов, соответственно, каждый из которых содержит четыре гена, кодирующие продукты, участвующие в сигнальном подпуте митоген-активируемой протеинкиназы (MAPK) (MAP2K1, RAF1, ELK1 и PIK3CA) ( Инжир.3). Эта общая подгруппа в сигнале GSEA сигнальных путей Ras, Ngf и Igf1 указывает на активацию этого компонента пути MAPK как на ключевое различие между опухолями p53 — и p53 + . (Отметим, что полный путь MAPK отображается как девятый набор в списке.)
Рис. 3.Передний край перекрытия для исследования p53. На этом графике показаны наборы генов ras , ngf и igf1 , коррелированные с P53 —, сгруппированные по их передним подмножествам, обозначенным темно-синим цветом.Общая подгруппа генов, видимая как темная вертикальная полоса, состоит из MAP2K1, PIK3CA, ELK1 и RAF1 и представляет собой часть пути MAPK.
Острые лейкозы. Затем мы попытались изучить острый лимфоидный лейкоз (ОЛЛ) и острый миелоидный лейкоз (ОМЛ) путем сравнения профилей экспрессии генов, которые мы ранее получили от 24 пациентов с ОЛЛ и 24 пациентов с ОМЛ ( 16).
Мы применили GSEA к цитогенетическим наборам генов (C 1 ), ожидая, что хромосомные полосы, показывающие обогащение в одном классе, вероятно, будут представлять области частых цитогенетических изменений при одной из двух лейкозов.Сравнение ALL> AML дало пять наборов генов ( Таблица 2), что может отражать частую амплификацию при ОЛЛ или делецию при ОМЛ. Действительно, все пять регионов легко интерпретируются с точки зрения современных знаний о лейкемии.
Полоса 5q31 соответствует известной цитогенетике ОМЛ. Делеции хромосомы 5q присутствуют у большинства пациентов с ОМЛ, причем критическая область локализована в 5q31 ( 17). Полоса 17q23 является участком известных генетических перестроек при миелоидных злокачественных новообразованиях ( 18).Полоса 13q14, содержащая локус RB, часто удаляется при AML, но редко при ALL ( 19). Наконец, полоса 6q21 содержит участок общей ломкости хромосом и обычно удаляется при гематологических злокачественных новообразованиях ( 20).
Интересно, что оставшаяся полоса с высокими показателями — 14q32. Эта полоса содержит локус тяжелой цепи Ig, который включает> 100 генов, экспрессируемых почти исключительно в лимфоидной линии. Обогащение 14q32 ALL, таким образом, отражает тканеспецифическую экспрессию в этой линии, а не хромосомную аномалию.
Реципрокный анализ (AML> ALL) не выявил значительно обогащенных полос, что, вероятно, отражает относительную редкость делеций при ALL ( 21). Таким образом, анализ наборов цитогенетических генов показывает, что GSEA способна идентифицировать хромосомные аберрации, общие для определенных подтипов рака.
Сравнение двух исследований рака легких. Цель GSEA — предоставить более надежный способ сравнения независимо полученных наборов данных экспрессии генов (возможно, полученных на разных платформах) и получить более согласованные результаты, чем анализ одного гена.Чтобы проверить надежность, мы повторно проанализировали данные двух недавних исследований рака легких, проведенных нашей собственной группой в Бостоне ( 22) и еще одна группа в Мичигане ( 23). Наша цель заключалась не в оценке результатов отдельных исследований, а в том, чтобы изучить, можно ли более эффективно выявить общие черты между наборами данных с помощью анализа набора генов, а не анализа одного гена.
Оба исследования определили профили экспрессии генов в образцах опухолей пациентов с аденокарциномами легких ( n = 62 для Бостона; n = 86 для Мичигана) и предоставили клинические результаты (классифицированные здесь как «хорошие» или «плохие») .Мы обнаружили, что никакие гены в обоих исследованиях не были сильно связаны с исходом на уровне значимости 5% после корректировки для проверки нескольких гипотез.
С точки зрения отдельных генов данные двух исследований мало общего. Традиционный подход заключается в сравнении генов, наиболее сильно коррелирующих с фенотипом. Мы определили набор генов S Boston как топ-100 генов, коррелирующих с плохим результатом в Бостонском исследовании, и аналогично S Michigan из Мичиганского исследования.Перекрытие крайне мало (12 общих генов) и едва ли является статистически значимым с тестом на перестановку ( P = 0,012). Когда мы добавили Стэнфордское исследование ( 24) с участием 24 аденокарцином, три набора данных имеют только один общий ген среди 100 лучших генов, коррелирующих с плохим результатом (рис. 5 и таблица 6, которые опубликованы в качестве вспомогательной информации на веб-сайте PNAS). Более того, из генов в перекрытиях не вытекает никаких четких общих тем, чтобы обеспечить биологическое понимание.
Затем мы исследовали, сможет ли GSEA выявить большее сходство между наборами данных по раку легких в Бостоне и Мичигане. Мы сравнили набор генов из одного набора данных, S Boston , со всем ранжированным списком генов из другого. Набор S Boston показывает сильное значительное обогащение данных Мичигана ( NES = 1,90, P <0,001). И наоборот, набор для неудачных исходов S Michigan обогащен данными Бостона ( NES = 2.13, P <0,001). Таким образом, GSEA может обнаружить сильный общий сигнал в данных о неблагоприятных исходах (рис. 6, который опубликован в качестве вспомогательной информации на веб-сайте PNAS).
Обнаружив, что GSEA может обнаруживать сходство между независимо полученными наборами данных, мы затем выяснили, может ли GSEA обеспечить биологическое понимание путем выявления важных функциональных наборов, коррелирующих с плохим исходом при раке легких. С этой целью мы выполнили GSEA по данным Бостона и Мичигана с каталогом функциональных наборов генов C 2 .Учитывая относительно слабые сигналы, обнаруженные обычным анализом одного гена в каждом исследовании, было неясно, будут ли обнаружены какие-либо значимые наборы генов с помощью GSEA. Тем не менее, мы определили ряд наборов генов, значимо коррелировавших с плохим исходом (FDR ≤ 0,25): 8 в данных Бостона и 11 в данных Мичигана ( Таблица 2). (В данных Стэнфорда не было генов или наборов генов, значимо коррелировавших с результатом, что, скорее всего, связано с меньшим количеством образцов и множеством отсутствующих значений в данных.)
Более того, в этих двух исследованиях наблюдается большое совпадение среди значительно обогащенных наборов генов. Примерно половина значимых наборов генов были общими для двух исследований, а несколько дополнительных, хотя и не идентичных, были явно связаны с одним и тем же биологическим процессом. В частности, мы обнаружили набор, активируемый теломеразой ( 25), два разных набора, связанных с синтезом тРНК, два разных набора, связанных с инсулином, и два разных набора, связанных с p53. Таким образом, в общей сложности 5 из 8 значимых наборов в Бостоне идентичны или связаны с 6 из 11 наборов в Мичигане.
Чтобы обеспечить более глубокое понимание, мы затем расширили анализ, включив в него наборы, выходящие за рамки тех, которые соответствуют критерию FDR ≤ 0,25. В частности, мы рассмотрели 20 наборов генов, набравших наибольшее количество баллов в каждом из трех исследований (60 наборов генов), и их соответствующие передовые подмножества, чтобы лучше понять лежащую в основе биологию в выборках с плохим исходом (таблица 4). Уже в районе пересечения Бостона и Мичигана мы увидели свидетельства теломеразы и реакции p-53, как отмечалось выше. Активация теломеразы считается ключевым аспектом патогенеза аденокарциномы легких и хорошо документирована как прогностический фактор неблагоприятного исхода при раке легких.
Во всех трех исследованиях возникают две дополнительные темы, связанные с быстрой клеточной пролиферацией и биосинтезом аминокислот (таблица 7, которая опубликована в качестве вспомогательной информации на веб-сайте PNAS):
Мы видим поразительные доказательства во всех трех исследованиях эффектов быстрой пролиферации клеток, включая наборы, связанные с активацией Ras и клеточным циклом, а также ответами на гипоксию, включая ангиогенез, гликолиз и метаболизм углеводов. Более трети наборов генов (23 из 60) связаны с такими процессами.Эти реакции наблюдались в микроокружении злокачественных опухолей, где усиленная пролиферация опухолевых клеток приводит к низким уровням кислорода и глюкозы ( 26). Ведущие подмножества ассоциированных значимых наборов генов включают гены реакции на гипоксию, такие как HIF1A, VEGF, CRK, PXN, EIF2B1, EIF2B2, EIF2S2, FADD, NFKB1, RELA, GADD45A, а также гены активации Ras / MAPK (HRAS, RAF1 и MAP2K1).
Мы находим убедительные доказательства одновременного присутствия повышенного биосинтеза аминокислот, передачи сигналов mTor и повышения регуляции набора генов, подавляемых как лишением аминокислот, так и лечением рапамицином ( 27).Это открытие подтверждают 17 наборов генов, связанных с метаболизмом аминокислот и нуклеотидов, иммунной модуляцией и передачей сигналов mTor . Основываясь на этих результатах, можно предположить, что лечение рапамицином может влиять на этот конкретный компонент сигнала о плохом исходе. Мы отмечаем, что существуют доказательства эффективности рапамицина в ингибировании роста и метастатического прогрессирования немелкоклеточного рака легкого у мышей и клеточных линий человека ( 28).
Наш анализ показывает, что мы обнаруживаем гораздо большую согласованность между тремя наборами данных о легких с помощью GSEA, чем с помощью анализа одного гена.Более того, мы лучше способны генерировать убедительные гипотезы для дальнейшего изучения. В частности, 40 из 60 наборов генов, набравших наибольшее количество баллов в этих трех исследованиях, дают последовательную картину основных биологических процессов в случаях плохого исхода.
Обсуждение
Традиционные стратегии анализа экспрессии генов сосредоточены на идентификации отдельных генов, которые демонстрируют различия между двумя интересующими состояниями. Несмотря на то, что они полезны, они не могут обнаружить биологические процессы, такие как метаболические пути, программы транскрипции и стрессовые реакции, которые распределены по всей сети генов и едва различимы на уровне отдельных генов.
Ранее мы ввели GSEA для анализа таких данных на уровне наборов генов. Первоначально этот метод использовался для обнаружения метаболических путей, измененных при диабете человека, а затем был применен для обнаружения процессов, участвующих в диффузной крупноклеточной лимфоме B ( 29), пути восприятия питательных веществ, участвующие в раке простаты ( 30), и при сравнении профилей экспрессии мыши и человека ( 31). В данной статье мы усовершенствовали оригинальный подход до чувствительного, надежного аналитического метода и инструмента с гораздо более широким применением, а также с большой базой данных наборов генов.GSEA может быть применен к другим наборам данных, таким как данные протеомики сыворотки, информация о генотипировании или профили метаболитов.
GSEA имеет ряд преимуществ по сравнению с методами с одним геном. Во-первых, он упрощает интерпретацию крупномасштабного эксперимента, выявляя пути и процессы. Вместо того, чтобы сосредотачиваться на генах с высокими показателями (которые могут быть плохо аннотированы и могут быть невоспроизводимы), исследователи могут сосредоточиться на наборах генов, которые имеют тенденцию быть более воспроизводимыми и более интерпретируемыми.Во-вторых, когда члены набора генов демонстрируют сильную взаимную корреляцию, GSEA может повысить отношение сигнал / шум и сделать возможным обнаружение умеренных изменений в отдельных генах. В-третьих, передовой анализ может помочь определить подмножества генов, чтобы прояснить результаты.
Недавно было разработано несколько других инструментов для анализа экспрессии генов с использованием информации о путях или онтологии, например, ( 32– 34). Большинство из них определяют, обогащается ли группа дифференциально экспрессируемых генов термином пути или онтологии, используя статистику перекрытия, такую как кумулятивное гипергеометрическое распределение.Отметим, что этот подход не может обнаружить результаты окислительного фосфорилирования, обсужденные выше ( P = 0,08, FDR = 0,50). GSEA отличается двумя важными аспектами. Во-первых, GSEA рассматривает все гены в эксперименте, а не только те, которые находятся выше произвольного порогового значения, с точки зрения кратности изменения или значимости. Во-вторых, GSEA оценивает значимость путем перестановки меток классов, что сохраняет корреляции ген-ген и, таким образом, обеспечивает более точную нулевую модель.
Однако реальная сила GSEA заключается в его гибкости.Мы создали исходную базу данных молекулярных сигнатур, состоящую из 1325 наборов генов, включая те, которые основаны на биологических путях, хромосомном положении, восходящих цис-мотивах, ответах на лечение лекарствами или профилях экспрессии в ранее созданных наборах данных микрочипов. Дополнительные наборы могут быть созданы с помощью генетических и химических возмущений, компьютерного анализа геномной информации и дополнительных биологических аннотаций. Кроме того, сам GSEA может быть использован для уточнения вручную выбранных путей и наборов путем определения передовых наборов, которые используются в различных наборах экспериментальных данных.По мере добавления таких наборов такие инструменты, как GSEA, помогут связать предыдущие знания с вновь созданными данными и тем самым помочь раскрыть коллективное поведение генов в состояниях здоровья и болезней.
Благодарности
Мы признательны за обсуждения или данные от D. Altshuler, N. Patterson, J. Lamb, X. Xie, J.-Ph. Брюне, С. Рамасвами, Ж.-П. Буркин, Б. Селлерс, Л. Стурла, К. Натт и Дж. К. Флорез и комментарии рецензентов.
Приложение: Математическое описание методов
Входы в GSEA.
Набор данных экспрессии D с N генами и k образцами.
Процедура ранжирования для составления Списка генов L . Включает корреляцию (или другую метрику ранжирования) и интересующий фенотип или профиль C . Мы используем только один зонд на каждый ген, чтобы предотвратить завышение статистики обогащения ( Вспомогательный текст ; см. Также Таблицу 8, которая опубликована в качестве вспомогательной информации на веб-сайте PNAS).
Показатель степени p для управления весом шага.
Независимо полученный набор генов S из N H генов (например, ., путь, цитогенетическая полоса или категория GO). В приведенном выше анализе мы использовали только наборы генов, содержащие не менее 15 членов, чтобы сосредоточиться на надежных сигналах (78% MSigDB) (таблица 3).
Enrichment Score ES (S).
Ранжируйте гены N в D с образованием L = { g 1 ,… , g N } согласно корреляции, r ( g j ) = r j , их профилей экспрессии с C .
Оцените долю генов в S («совпадения»), взвешенную по их корреляции, и долю генов, отсутствующих в S («промахи»), присутствующих до данной позиции и в L .
$$ mathtex $$$$ mathtex $$ [1] $$ mathtex $$$$ mathtex $$
ES — максимальное отклонение от нуля P попадание — P промах .Для произвольно распределенного S, ES ( S ) будет относительно небольшим, но если он сосредоточен в верхней или нижней части списка или иным образом распределен неслучайно, то ES ( S ) будет соответственно высокая. Когда p = 0, ES (S) сводится к стандартной статистике Колмогорова – Смирнова; когда p = 1, мы взвешиваем гены в S по их корреляции с C , нормализованной суммой корреляций по всем генам в S .Мы устанавливаем p = 1 для примеров в этой статье. (См. Рис. 7, который опубликован в качестве вспомогательной информации на веб-сайте PNAS.)
Оценка значимости. Мы оцениваем значимость наблюдаемого ES , сравнивая его с набором баллов ES NULL , вычисленных со случайно назначенными фенотипами.
Случайным образом назначьте исходные метки фенотипа образцам, измените порядок генов и повторно вычислите ES ( S ).
Повторите шаг 1 для 1000 перестановок и создайте гистограмму соответствующих показателей обогащения ES NULL .
Оцените номинальное значение P для S из ES NULL , используя положительную или отрицательную часть распределения, соответствующую знаку наблюдаемого ES ( S ).
Проверка множественных гипотез.
Определите ES ( S ) для каждого набора генов в коллекции или базе данных.
Для каждых S и 1000 фиксированных перестановок π меток фенотипа переупорядочить гены в L и определить ES ( S , π).
Скорректировать вариации в размере набора генов. Нормализуйте ES ( S , π) и наблюдаемое ES ( S ), отдельно изменив масштаб положительных и отрицательных оценок путем деления на среднее значение ES ( S , π), чтобы получить нормализованные оценки NES ( S , π) и NES ( S ) (см. Вспомогательный текст ).
Вычислить FDR. Контролируйте соотношение ложноположительных результатов к общему количеству наборов генов, достигающих фиксированного уровня значимости, отдельно для положительных (отрицательных) NES ( S ) и NES ( S , π).
Создайте гистограмму всех NES ( S , π) по всем S и π. Используйте это нулевое распределение для вычисления значения FDR q для данного NES ( S ) = NES * ≥ 0. FDR — это соотношение процентного содержания всех ( S , π) с NES ( S , π) ≥ 0, у которых NES ( S , π) ≥ NES *, деленное на процент наблюдаемых S с NES ( S ) ≥ 0, чей NES ( S ) ≥ NES *, и аналогично, если NES ( S ) = NES * ≤ 0.
Сноски
↵k Кому может быть адресована корреспонденция.Электронная почта: lander {at} broad.mit.edu или mesirov {at} broad.mit.edu.
↵b A.S. и П. внес равный вклад в эту работу.
Вклад авторов: A.S., P.T., V.K.M., E.S.L. и J.P.M. спланированное исследование; A.S., P.T., V.K.M., E.S.L. и J.P.M. проведенное исследование; A.S., P.T., V.K.M., S.M., E.S.L. и J.P.M. внесены новые реагенты / аналитические инструменты; A.S., P.T., V.K.M., B.L.E., M.A.G., T.R.G., E.S.L. и J.P.M. проанализированные данные; A.S., P.T., V.K.M., E.S.L. и J.ВЕЧЕРА. написал статью; и A.P. и S.L.P. предоставленные данные.
Сокращения: ОЛЛ, острый лимфолейкоз; ОМЛ, острый миелоидный лейкоз; ES , оценка обогащения; FDR — коэффициент ложного обнаружения; GSEA, Анализ обогащения набора генов; MAPK, митоген-активированная протеинкиназа; MSigDB, База данных молекулярных сигнатур; NES, нормализованная оценка обогащения.
См. Комментарий на стр. 15278.
- Авторские права © 2005, Национальная академия наук
Доступно бесплатно в Интернете через опцию открытого доступа PNAS.